Differential discovery analysis
Timothy Keyes
2024-08-25
Source:vignettes/differential-discovery-analysis.Rmd
differential-discovery-analysis.RmdAfter clusters are identified, many cytometrists want to use statistical tools to rigorously quantify which clusters(s) in their dataset associate with a particular experimental or clinical variable.
Such analyses are often grouped under the umbrella term
differential discovery analysis and include both
comparing the relative size of clusters between experimental
conditions (differential abundance analysis; DAA) as
well as comparing marker expression patterns of clusters between
experimental conditions (differential expression analysis;
DEA). tidytof provides the
tof_analyze_abundance() and
tof_analyze_expression() verbs for differential abundance
and differential expression analyses, respectively.
Accessing the data for this vignette
To demonstrate how to use these verbs, we’ll first download a dataset originally collected for the development of the CITRUS algorithm. These data are available in the HDCytoData package, which is available on Bioconductor and can be downloaded with the following command:
if (!requireNamespace("BiocManager", quietly = TRUE)) {
install.packages("BiocManager")
}
BiocManager::install("HDCytoData")To load the CITRUS data into our current R session, we can call a
function from the HDCytoData, which will provide it to us
in a format from the {flowCore} package (called a
“flowSet”). To convert this into a tidy tibble, we can use
tidytof built-in method for converting flowCore objects
into tof_tbl’s .
citrus_raw <- HDCytoData::Bodenmiller_BCR_XL_flowSet()
citrus_data <-
citrus_raw |>
as_tof_tbl(sep = "_")Thus, we can see that citrus_data is a
tof_tbl with 172791 cells (one in each row) and 39 pieces
of information about each cell (one in each column).
We can also extract some metadata from the raw data and join it with
our single-cell data using some functions from the
tidyverse:
citrus_metadata <-
tibble(
file_name = as.character(flowCore::pData(citrus_raw)[[1]]),
sample_id = 1:length(file_name),
patient = stringr::str_extract(file_name, "patient[:digit:]"),
stimulation = stringr::str_extract(file_name, "(BCR-XL)|Reference")
) |>
mutate(
stimulation = if_else(stimulation == "Reference", "Basal", stimulation)
)
citrus_metadata |>
head()
#> # A tibble: 6 × 4
#> file_name sample_id patient stimulation
#> <chr> <int> <chr> <chr>
#> 1 PBMC8_30min_patient1_BCR-XL.fcs 1 patient1 BCR-XL
#> 2 PBMC8_30min_patient1_Reference.fcs 2 patient1 Basal
#> 3 PBMC8_30min_patient2_BCR-XL.fcs 3 patient2 BCR-XL
#> 4 PBMC8_30min_patient2_Reference.fcs 4 patient2 Basal
#> 5 PBMC8_30min_patient3_BCR-XL.fcs 5 patient3 BCR-XL
#> 6 PBMC8_30min_patient3_Reference.fcs 6 patient3 BasalThus, we now have sample-level information about which patient each sample was collected from and which stimulation condition (“Basal” or “BCR-XL”) each sample was exposed to before data acquisition.
Finally, we can join this metadata with our single-cell
tof_tbl to obtain the cleaned dataset.
citrus_data <-
citrus_data |>
left_join(citrus_metadata, by = "sample_id")After these data cleaning steps, we now have
citrus_data, a tof_tbl containing cells
collected from 8 patients. Specifically, 2 samples were taken from each
patient: one in which the cells’ B-cell receptors were stimulated
(BCR-XL) and one in which they were not (Basal). In
citrus_data, each cell’s patient of origin is stored in the
patient column, and each cell’s stimulation condition is
stored in the stimulation column. In addition, the
population_id column stores information about cluster
labels that were applied to each cell using a combination of FlowSOM
clustering and manual merging (for details, run
?HDCytoData::Bodenmiller_BCR_XL in the R console).
Differential abundance analysis using
tof_analyze_abundance()
We might wonder if there are certain clusters that expand or deplete
within patients between the two stimulation conditions described above -
this is a question that requires differential abundance analysis (DAA).
tidytof’s tof_analyze_abundance() verb
supports the use of 3 statistical approaches for performing DAA:
diffcyt, generalized-linear mixed modeling (GLMMs), and simple t-tests.
Because the setup described above uses a paired design and only has 2
experimental conditions of interest (Basal vs. BCR-XL), we can use the
paired t-test method:
daa_result <-
citrus_data |>
tof_analyze_abundance(
cluster_col = population_id,
effect_col = stimulation,
group_cols = patient,
test_type = "paired",
method = "ttest"
)
daa_result
#> # A tibble: 8 × 8
#> population_id p_val p_adj significant t df mean_diff mean_fc
#> <chr> <dbl> <dbl> <chr> <dbl> <dbl> <dbl> <dbl>
#> 1 1 0.000924 0.00535 "*" -5.48 7 -0.00743 0.644
#> 2 2 0.00623 0.0166 "*" -3.86 7 -0.0156 0.674
#> 3 3 0.0235 0.0314 "*" -2.88 7 -0.0638 0.849
#> 4 4 0.0235 0.0314 "*" 2.88 7 0.0832 1.38
#> 5 5 0.0116 0.0232 "*" 3.39 7 0.00246 1.08
#> 6 6 0.371 0.371 "" -0.955 7 -0.0168 0.919
#> 7 7 0.00134 0.00535 "*" 5.14 7 0.0202 1.14
#> 8 8 0.236 0.270 "" -1.30 7 -0.00228 0.901Based on this output, we can see that 6 of our 8 clusters have statistically different abundance in our two stimulation conditions. Using tidytof easy integration with tidyverse packages, we can use this result to visualize the fold-changes of each cluster (within each patient) in the BCR-XL condition compared to the Basal condition using ggplot2:
plot_data <-
citrus_data |>
mutate(population_id = as.character(population_id)) |>
left_join(
select(daa_result, population_id, significant, mean_fc),
by = "population_id"
) |>
dplyr::count(patient, stimulation, population_id, significant, mean_fc, name = "n") |>
group_by(patient, stimulation) |>
mutate(prop = n / sum(n)) |>
ungroup() |>
pivot_wider(
names_from = stimulation,
values_from = c(prop, n),
) |>
mutate(
diff = `prop_BCR-XL` - prop_Basal,
fc = `prop_BCR-XL` / prop_Basal,
population_id = fct_reorder(population_id, diff),
direction =
case_when(
mean_fc > 1 & significant == "*" ~ "increase",
mean_fc < 1 & significant == "*" ~ "decrease",
TRUE ~ NA_character_
)
)
significance_data <-
plot_data |>
group_by(population_id, significant, direction) |>
summarize(diff = max(diff), fc = max(fc)) |>
ungroup()
plot_data |>
ggplot(aes(x = population_id, y = fc, fill = direction)) +
geom_violin(trim = FALSE) +
geom_hline(yintercept = 1, color = "red", linetype = "dotted", size = 0.5) +
geom_point() +
geom_text(
aes(x = population_id, y = fc, label = significant),
data = significance_data,
size = 8,
nudge_x = 0.2,
nudge_y = 0.06
) +
scale_x_discrete(labels = function(x) str_c("cluster ", x)) +
scale_fill_manual(
values = c("decrease" = "#cd5241", "increase" = "#207394"),
na.translate = FALSE
) +
labs(
x = NULL,
y = "Abundance fold-change (stimulated / basal)",
fill = "Effect",
caption = "Asterisks indicate significance at an adjusted p-value of 0.05"
)
#> Warning: Using `size` aesthetic for lines was deprecated in ggplot2 3.4.0.
#> ℹ Please use `linewidth` instead.
#> This warning is displayed once every 8 hours.
#> Call `lifecycle::last_lifecycle_warnings()` to see where this warning was
#> generated.
Importantly, the output of tof_analyze_abundance depends
slightly on the underlying statistical method being used, and details
can be found in the documentation for each
tof_analyze_abundance_* function family member:
tof_analyze_abundance_diffcyttof_analyze_abundance_glmmtof_analyze_abundance_ttest
Differential expression analysis with
tof_analyze_expression()
Similarly, suppose we’re interested in how intracellular signaling
proteins change their expression levels between our two stimulation
conditions in each of our clusters. This is a Differential Expression
Analysis (DEA) and can be performed using tidytof’s
tof_analyze_expression verb. As above, we can use paired
t-tests with multiple-hypothesis correction to to test for significant
differences in each cluster’s expression of our signaling markers
between stimulation conditions.
signaling_markers <-
c(
"pNFkB_Nd142", "pStat5_Nd150", "pAkt_Sm152", "pStat1_Eu153", "pStat3_Gd158",
"pSlp76_Dy164", "pBtk_Er166", "pErk_Er168", "pS6_Yb172", "pZap70_Gd156"
)
dea_result <-
citrus_data |>
tof_preprocess(channel_cols = any_of(signaling_markers)) |>
tof_analyze_expression(
method = "ttest",
cluster_col = population_id,
marker_cols = any_of(signaling_markers),
effect_col = stimulation,
group_cols = patient,
test_type = "paired"
)
dea_result |>
head()
#> # A tibble: 6 × 9
#> population_id marker p_val p_adj significant t df mean_diff mean_fc
#> <chr> <chr> <dbl> <dbl> <chr> <dbl> <dbl> <dbl> <dbl>
#> 1 1 pS6_Y… 7.58e-8 2.12e-6 * 22.9 7 2.56 4.31
#> 2 2 pS6_Y… 1.16e-7 2.12e-6 * 21.6 7 2.13 2.49
#> 3 3 pBtk_… 1.32e-7 2.12e-6 * -21.2 7 -0.475 0.289
#> 4 7 pBtk_… 1.18e-7 2.12e-6 * -21.5 7 -0.518 0.286
#> 5 8 pBtk_… 1.30e-7 2.12e-6 * -21.2 7 -0.516 0.324
#> 6 4 pBtk_… 7.85e-7 1.05e-5 * -16.3 7 -0.462 0.296While the output of tof_analyze_expression() also
depends on the underlying test being used, we can see that the result
above looks relatively similar to the output from
tof_analyze_abundance(). Above, the output is a tibble in
which each row represents the differential expression results from a
single cluster-marker pair - for example, the first row represents the
difference in expression of pS6 in cluster 1 between the BCR-XL and
Basal conditions. Each row includes the raw p-value and
multiple-hypothesis-corrected p-value for each cluster-marker pair.
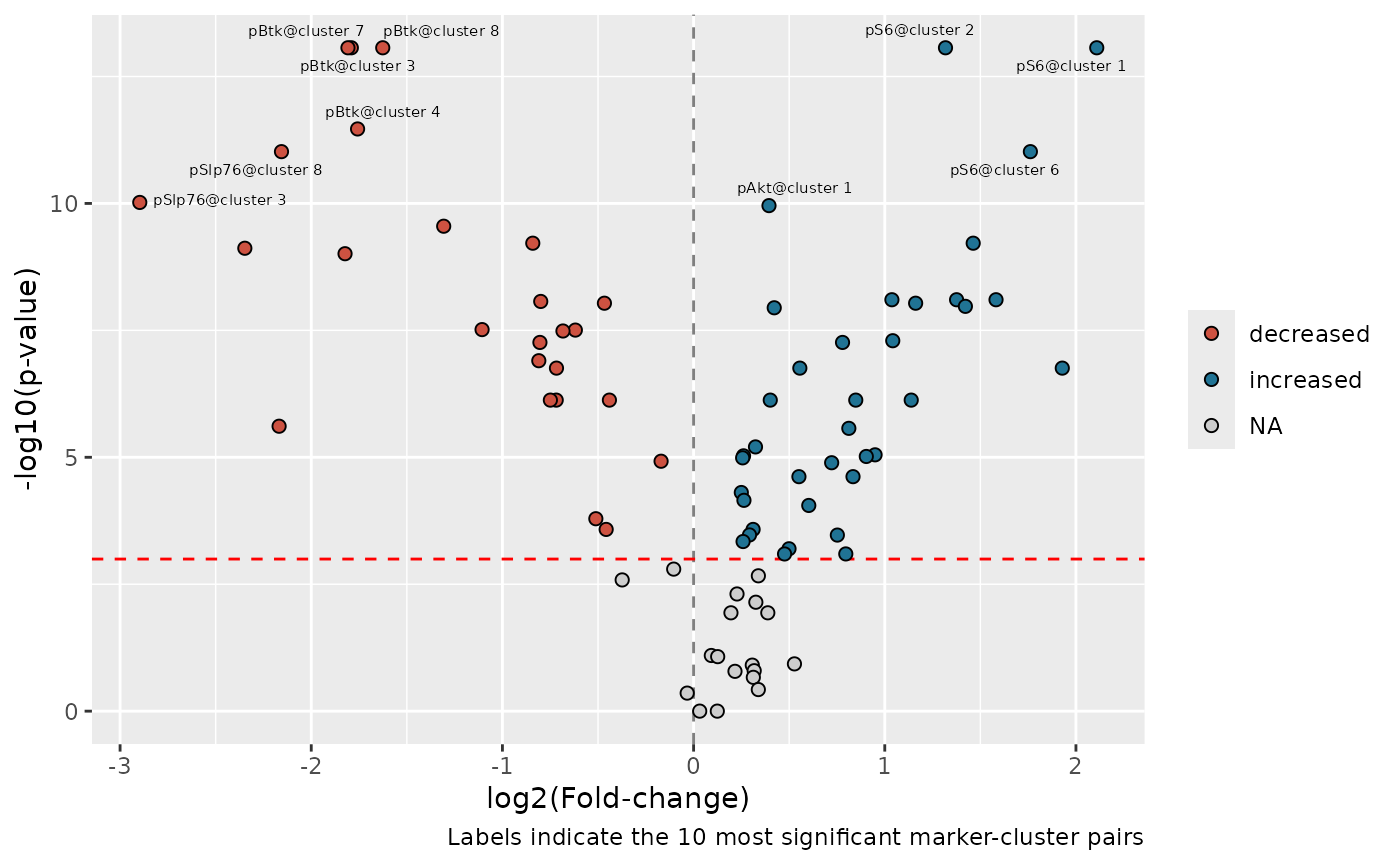
This result can be used to make a volcano plot to visualize the results for all cluster-marker pairs:
volcano_data <-
dea_result |>
mutate(
log2_fc = log(mean_fc, base = 2),
log_p = -log(p_adj),
significance =
case_when(
p_adj < 0.05 & mean_fc > 1 ~ "increased",
p_adj < 0.05 & mean_fc < 1 ~ "decreased",
TRUE ~ NA_character_
),
marker =
str_extract(marker, ".+_") |>
str_remove("_"),
pair = str_c(marker, str_c("cluster ", population_id), sep = "@")
)
volcano_data |>
ggplot(aes(x = log2_fc, y = log_p, fill = significance)) +
geom_vline(xintercept = 0, linetype = "dashed", color = "gray50") +
geom_hline(yintercept = -log(0.05), linetype = "dashed", color = "red") +
geom_point(shape = 21, size = 2) +
ggrepel::geom_text_repel(
aes(label = pair),
data = slice_head(volcano_data, n = 10L),
size = 2
) +
scale_fill_manual(
values = c("decreased" = "#cd5241", "increased" = "#207394"),
na.value = "#cdcdcd"
) +
labs(
x = "log2(Fold-change)",
y = "-log10(p-value)",
fill = NULL,
caption = "Labels indicate the 10 most significant marker-cluster pairs"
)
As above, details can be found in the documentation for each
tof_analyze_expression_* function family member:
tof_analyze_expression_diffcyttof_analyze_expression_lmmtof_analyze_expression_ttest
Session info
sessionInfo()
#> R version 4.4.1 (2024-06-14)
#> Platform: x86_64-pc-linux-gnu
#> Running under: Ubuntu 22.04.4 LTS
#>
#> Matrix products: default
#> BLAS: /usr/lib/x86_64-linux-gnu/openblas-pthread/libblas.so.3
#> LAPACK: /usr/lib/x86_64-linux-gnu/openblas-pthread/libopenblasp-r0.3.20.so; LAPACK version 3.10.0
#>
#> locale:
#> [1] LC_CTYPE=C.UTF-8 LC_NUMERIC=C LC_TIME=C.UTF-8
#> [4] LC_COLLATE=C.UTF-8 LC_MONETARY=C.UTF-8 LC_MESSAGES=C.UTF-8
#> [7] LC_PAPER=C.UTF-8 LC_NAME=C LC_ADDRESS=C
#> [10] LC_TELEPHONE=C LC_MEASUREMENT=C.UTF-8 LC_IDENTIFICATION=C
#>
#> time zone: UTC
#> tzcode source: system (glibc)
#>
#> attached base packages:
#> [1] stats4 stats graphics grDevices utils datasets methods
#> [8] base
#>
#> other attached packages:
#> [1] HDCytoData_1.24.0 flowCore_2.16.0
#> [3] SummarizedExperiment_1.34.0 Biobase_2.64.0
#> [5] GenomicRanges_1.56.1 GenomeInfoDb_1.40.1
#> [7] IRanges_2.38.1 S4Vectors_0.42.1
#> [9] MatrixGenerics_1.16.0 matrixStats_1.3.0
#> [11] ExperimentHub_2.12.0 AnnotationHub_3.12.0
#> [13] BiocFileCache_2.12.0 dbplyr_2.5.0
#> [15] BiocGenerics_0.50.0 forcats_1.0.0
#> [17] tidyr_1.3.1 ggplot2_3.5.1
#> [19] stringr_1.5.1 dplyr_1.1.4
#> [21] tidytof_0.99.8
#>
#> loaded via a namespace (and not attached):
#> [1] jsonlite_1.8.8 shape_1.4.6.1 magrittr_2.0.3
#> [4] farver_2.1.2 rmarkdown_2.28 fs_1.6.4
#> [7] zlibbioc_1.50.0 ragg_1.3.2 vctrs_0.6.5
#> [10] memoise_2.0.1 htmltools_0.5.8.1 S4Arrays_1.4.1
#> [13] curl_5.2.1 SparseArray_1.4.8 sass_0.4.9
#> [16] parallelly_1.38.0 bslib_0.8.0 htmlwidgets_1.6.4
#> [19] desc_1.4.3 lubridate_1.9.3 cachem_1.1.0
#> [22] igraph_2.0.3 mime_0.12 lifecycle_1.0.4
#> [25] iterators_1.0.14 pkgconfig_2.0.3 Matrix_1.7-0
#> [28] R6_2.5.1 fastmap_1.2.0 GenomeInfoDbData_1.2.12
#> [31] future_1.34.0 digest_0.6.37 colorspace_2.1-1
#> [34] AnnotationDbi_1.66.0 textshaping_0.4.0 RSQLite_2.3.7
#> [37] labeling_0.4.3 filelock_1.0.3 cytolib_2.16.0
#> [40] fansi_1.0.6 yardstick_1.3.1 timechange_0.3.0
#> [43] httr_1.4.7 polyclip_1.10-7 abind_1.4-5
#> [46] compiler_4.4.1 bit64_4.0.5 withr_3.0.1
#> [49] doParallel_1.0.17 viridis_0.6.5 DBI_1.2.3
#> [52] highr_0.11 ggforce_0.4.2 MASS_7.3-60.2
#> [55] lava_1.8.0 rappdirs_0.3.3 DelayedArray_0.30.1
#> [58] tools_4.4.1 future.apply_1.11.2 nnet_7.3-19
#> [61] glue_1.7.0 grid_4.4.1 generics_0.1.3
#> [64] recipes_1.1.0 gtable_0.3.5 tzdb_0.4.0
#> [67] class_7.3-22 data.table_1.15.4 hms_1.1.3
#> [70] tidygraph_1.3.1 utf8_1.2.4 XVector_0.44.0
#> [73] ggrepel_0.9.5 BiocVersion_3.19.1 foreach_1.5.2
#> [76] pillar_1.9.0 RcppHNSW_0.6.0 splines_4.4.1
#> [79] tweenr_2.0.3 lattice_0.22-6 survival_3.6-4
#> [82] bit_4.0.5 RProtoBufLib_2.16.0 tidyselect_1.2.1
#> [85] Biostrings_2.72.1 knitr_1.48 gridExtra_2.3
#> [88] xfun_0.47 graphlayouts_1.1.1 hardhat_1.4.0
#> [91] timeDate_4032.109 stringi_1.8.4 UCSC.utils_1.0.0
#> [94] yaml_2.3.10 evaluate_0.24.0 codetools_0.2-20
#> [97] ggraph_2.2.1 tibble_3.2.1 BiocManager_1.30.24
#> [100] cli_3.6.3 rpart_4.1.23 systemfonts_1.1.0
#> [103] munsell_0.5.1 jquerylib_0.1.4 Rcpp_1.0.13
#> [106] globals_0.16.3 png_0.1-8 parallel_4.4.1
#> [109] pkgdown_2.1.0 gower_1.0.1 readr_2.1.5
#> [112] blob_1.2.4 listenv_0.9.1 glmnet_4.1-8
#> [115] viridisLite_0.4.2 ipred_0.9-15 scales_1.3.0
#> [118] prodlim_2024.06.25 purrr_1.0.2 crayon_1.5.3
#> [121] rlang_1.1.4 KEGGREST_1.44.1